IN A NUTSHELL
1. About This Work
In the course of infecting their hosts, pathogenic bacteria secrete numerous effectors: bacterial proteins that pervert host cell biology. Many Gram-negative bacteria, including context-dependent human pathogens, use a type IV secretion system (T4SS) to translocate effectors directly into the cytosol of host cells. Various type IV secreted effectors (T4SEs) have been experimentally validated to play crucial roles in virulence by manipulating host cell gene expression and other processes. Consequently, the identification of novel effector proteins is an important step in increasing our understanding of host-pathogen interactions and bacterial pathogenesis.
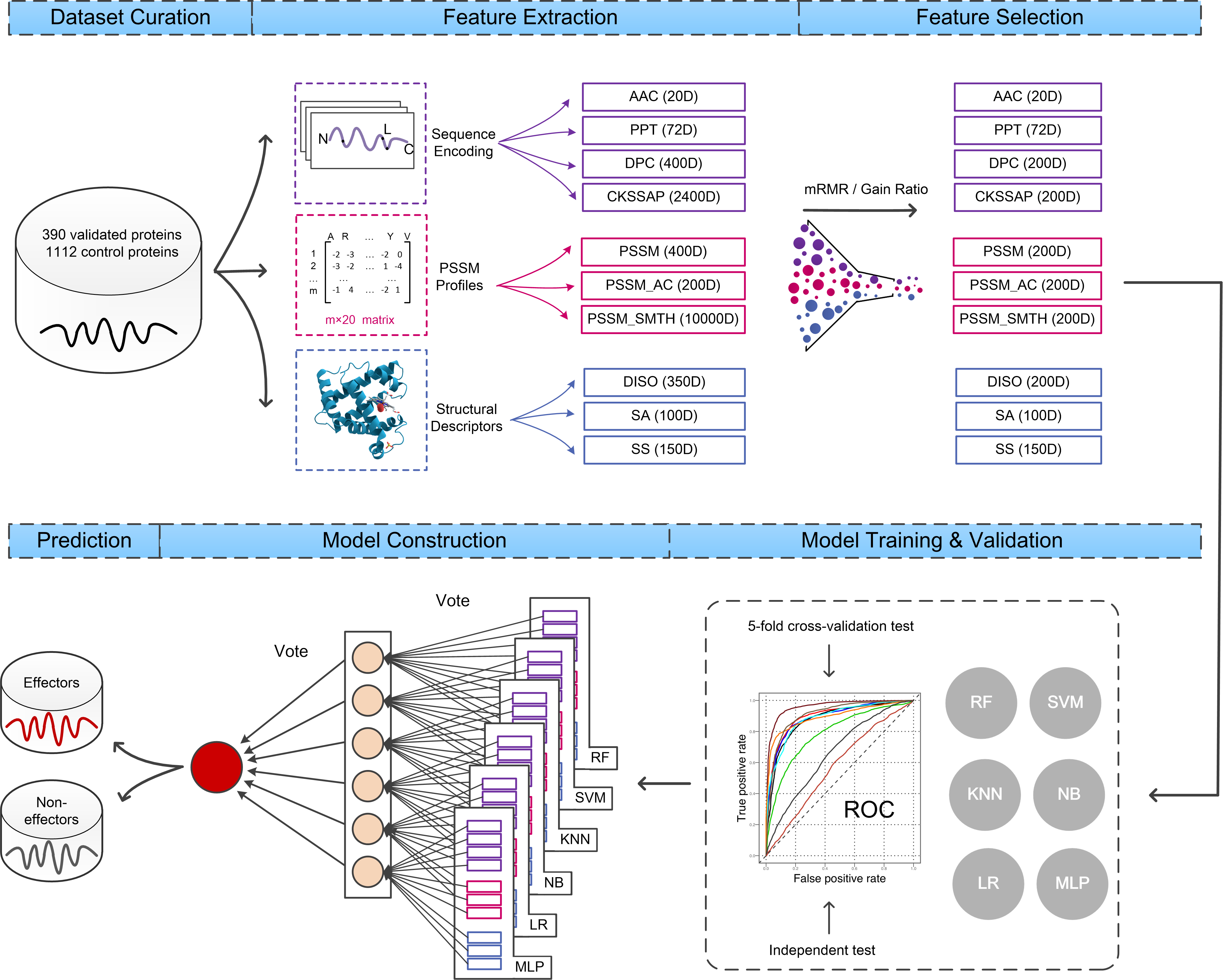
In this study we developed a state-of-the-art T4SE predictor by conducting a comprehensive performance evaluation of different machine learning algorithms along with a detailed analysis of single and multi-feature selections.
2. Key Points Of Our Work
- This work systematically trained and compared six commonly used machine learning models, namely: NB, KNN, LR, RF, SVM and MLP, for the identification of type IV secretion system effectors (T4SEs) using ten different types of selected features.
- Our study showed that (1) including different but complementary features generally enhanced the predictive performance of T4SEs; (2) ensemble models obtained by integrating individual single-feature models exhibited a significantly improved predictive performance. The majority voting strategy enables the ensemble models to achieve the most stable and accurate classification performance.
- We thus proposed and built a new ensemble model, Bastion4, to further improve the performance in predicting effector proteins of the type IV secretion system. Independent tests demonstrate that the ensemble models outperform all current predictors of types IV secretion systems. Bastion4 is publicly accessible at http://bastion4.erc.monash.edu/.
- Genome-wide prediction of T4SEs provided important insights into the distribution of T4SEs in three bacterial pathogens, providing a valuable compendium of novel T4SEs that can be further validated by genetic and biochemical experiments.
3. Corresponding Authors
Trevor Lithgow, Biomedicine Discovery Institute and Department of Microbiology, Monash University, Melbourne, Victoria 3800, Australia. Tel: +61-3-9902-9217 Fax: +61-3-9905-3726 Email: Trevor.Lithgow@monash.edu
Jiangning Song, Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Victoria 3800, Australia. Tel: +61-3-9902-9304 Email: Jiangning.Song@monash.edu
DATASETS
1. Construction of the Training Dataset
The input dataset consisted of two parts, the training dataset and the independent dataset. We constructed the training dataset by extracting known T4SEs from different datasets described in the literature. Specifically, 347 T4SE sequences were extracted from the T4SEpre dataset constructed by Wang et al.. Then, 340 effectors including 30 IVA proteins and 310 IVB proteins were acquired from Zou et al.. Subsequently, we added 120 proteins given by Burstein et al.. For the negative training set, we chose the entire set of 1132 non-effectors in Zou et al.. After forming the preliminary dataset, CD-HIT was used to remove highly homologous sequences (at the 60% sequence identity) to reduce sequence redundancy, which may otherwise lead to a potential bias in the trained models. The final training dataset contained 390 152 positive and 1112 negative sequences.
2. Construction of the Independent Test Dataset
In order to evaluate the model performance in comparison with existing T4SE prediction tools, we generated an independent dataset containing both positive and negative samples. For the former, 43 positive samples were acquired from the UniProt Database and Meyer et al., and for the latter, 150 samples from the dataset of Vibrio parahaemolyticus serotype O3:K6 (strain RIMD 158 2210633). After removal of duplicate samples, which appear in our training set and the datasets used by the existing T4SE predictors, we obtained a final independent dataset made up of 30 positive and 150 negative samples.
ONLINE WEB SERVER
1. Bastion4
As an implementation of the methodology presented here, we developed an online webserver of Bastion4 for characterizing protein sequences of interest, freely accessible at http://bastion4.erc.monash.edu.au. The Bastion4 webserver was configured on the cloud computing facility provided by the Monash University e-Research Centre and equipped with four instances, dual-core processors, 16GB of memory and a 200 GB hard disk. This configuration can be further extended to meet future requirements. The server used Perl in combination with J2EE to provide the web service. As input, one or more protein sequences can be submitted to the online webserver. Users are able to select and combine any individual models to customize an ensemble predictor. Additionally, the webpage presents an example of amino acid sequences in FASTA format, and a particular ensemble model is chosen as the default. The computational time of the Bastion4 server to process a submitted sequence depends not only on the length of the submitted sequence but also considerably on the choice of selected models.
2. Using Bastion4
Bastion4 is an online server with a user-friendly interface and few parameters, therefore it is easy to use. All you need to do is to fill the input box and select the model you want to use. The prediction job will be put into the queue system. All the jobs will be executed by Bastion4 server successively. After your job is finished, you will receive an e-mail with a url of your job result.
2.1 Input Formats
Two types of input are accepted by Bastion4: sequences in FASTA format (recommended) and raw sequences.
For sequences in FASTA format, you can input as follows:
Also, the following input (which is the original formats downloaded from Uniprot database)
will be formated (without line break inside the sequence) as:
For the raw sequences, you can input as follows:
which will be formated by Bastion4 as follows:
2.2 Model used to custom Bastion4 ensemble predictor
Three types of sequence-encoding models (with 10 specific models) are available in Bastion4. In general, selecting only local-sequence-encoding based models enables high-speed predictions but at the expense of a reduced prediction accuracy. In contrast, global sequence-encoding or structural-descriptor-encoding based models can provide an improved predictive performance, but suffer from increased computational complexity and time consumption. Users are advised to be aware of the compromise between computational efficiency and prediction accuracy when selecting models. A job submission with the default settings will take the Bastion4 server approximately 10 minutes to process and accomplish the job of a typical protein sequence with 500 amino acid residues.
2.3 Input limits
1. The length of each submited sequence should be in the range of 50 and 5000.
2. Since T4SE prediction is a time-consuming job, especially when you select Global Sequence Encoding Based Models or Sequence functional Encoding Based Models, the max number of submited sequences each time should be no more than 50.